近日,我院孙世刚院士、王宇成副教授在非贵金属催化剂活性位点的精准表征研究中取得重要进展。相关成果以 “Identifying high-spin hydroxyl-coordinated Fe3+N4 as the active centre for acidic oxygen reduction using molecular model catalysts”为题在线发表于Nature Catalysis.
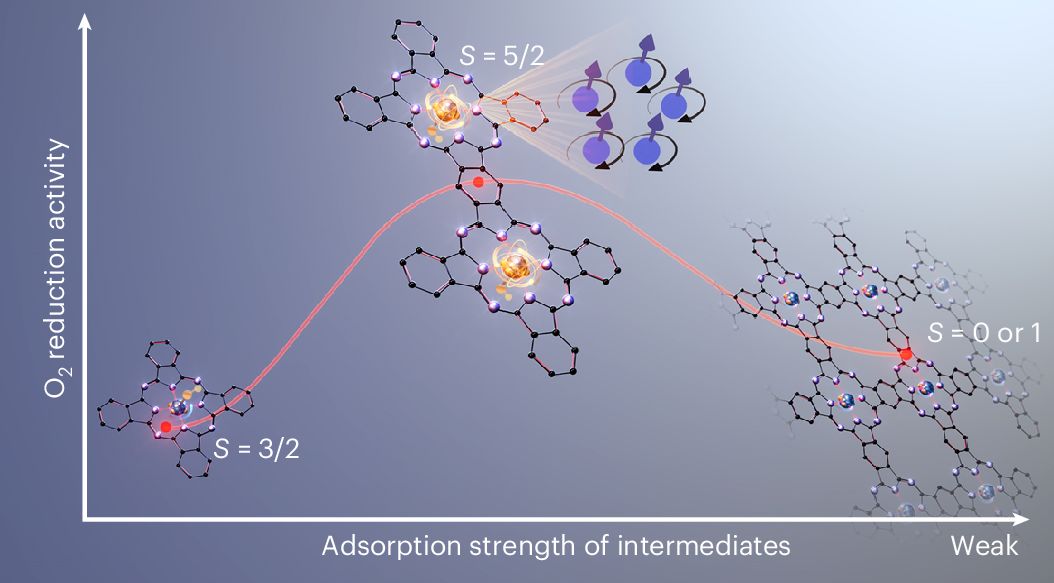
Fe–N–C被认为是最有前景的替代贵金属铂的非贵金属氧还原(ORR)电催化剂,但其活性中心的电子结构一直不明确。本研究合成了一种共轭桥联酞菁铁二聚体模型催化剂(FePc=FePc),通过原位与非原位X射线吸收、质谱、穆斯堡尔谱、电子顺磁共振、磁学测量等表征,精确鉴定出活性位点为OH–Fe3+N4 (S=5/2)。进一步,通过对比五种不同共轭分子催化剂的结构、物理性质和电化学活性,研究发现:FePc=FePc中的OH–Fe3+N4(S=5/2)位点表现出适宜的羟基吸附强度,可实现快速的ORR;而在单体或非共轭二聚体中,活性位点为OH–Fe3+N4(S=3/2)态,吸附能过强;聚合态酞菁铁则由35%的S=5/2态与65%的Fe2+N4(S=0或1)态混合组成,整体吸附能偏弱。结果表明,吸附能过强或过弱,均不利于ORR。
理论计算表明,铁中心与共轭碳平面之间的π-d相互作用对铁的自旋态具有决定性影响。从单体到共轭二聚体的转变过程中,增强的π-d相互作用会降低铁的氧化态,导致d轨道分裂能减小,使自旋态从中自旋态(S=3/2)转变为高自旋态(S=5/2)。而当共轭体系进一步延伸至聚合态酞菁铁时,过强的π-d相互作用会使其配位构型从四方锥形转变为平面四方,增大d轨道分裂能,使自旋态转变为中自旋态(S=1)。
更重要的是,从分子模型催化剂得出的结论似乎也适用于热解催化剂。该研究深化了对活性位点的理解,为精准设计ORR及其他反应的单原子催化剂提供了理论依据,对催化领域具有广泛意义。
该工作在王宇成副教授和孙世刚院士的共同指导下完成。我院博士后赵匡民博士、海南大学吴道雄副研究员和密歇根大学Wu Wen-kun博士为共同第一作者;聂佳宝博士完成理论计算;李广博士完成红外表征;时海燕工程师完成了穆斯堡尔谱表征;华东师范大学耿福山老师完成电子顺磁共振分析;中科院高能物理所张静研究员和黄换副研究员完成了同步辐射X射线吸收谱的表征与分析;嘉庚实验室黄声超工程师完成原子力显微镜的表征与分析;周志有教授为全文修改提供了宝贵意见。研究工作得到国家重点研发计划(2023YFA1509000),国家自然科学基金创新研究群体(22021001)、面上项目(22179116)、基础科学中心(22288102),中央高校基本科研业务费(20720220017)的资助。
论文链接: https://www.nature.com/articles/s41929-025-01324-7

